Barcoding and gRNA Probes
Compiled on September 16, 2023
Source:vignettes/articles/barcoding.Rmd
barcoding.RmdThis tutorial will demonstrate how to use karyotapR to
parse and count barcoded and gRNA reads using the specialty probes in
the KaryoTap Tapestri panels. These barcodes and gRNAs are integrated
into the cell’s genome using lentivirus and are amplified by specially
designed probes specific to the vectors. Details on the probe design can
be found in the KaryoTap publication, and details on the usage of the
functions here can be found in the package documentation and reference
on this site.
The procedures for counting barcoded reads and gRNA reads are largely identical, except for pointing the function to the correct probe. Since gRNAs sequences essentially act as barcodes, we’re going to refer to both barcodes and gRNAs as barcodes here.
Barcode sequences are extracted from the aligned reads stored in the
.bam file that is output as part of the Tapestri Pipeline.
We’ll use a dataset (experiment 4) from the KaryoTap paper as an
example. The .bam file used in this tutorial can be
downloaded from the SRA repository.
The file needs a corresponding .bai index file as well. If
you did not retrieve it from the Pipeline output, you can generate a new
one (see the countBarcodedReads() documentation).
To identify the barcoded sequences within the reads, we need a lookup table containing the barcode ID (whatever you want the name to appear as in the result) and the corresponding nucleotide sequences for the barcode.
grna.lookup <- data.frame(ids = c("gRNA1", "gRNA2"), sequences = c("ACGGAGGCTAAGCGTCGCAA",
"ACTCTTGCTGTGGCATTTTC"))Barcoded reads are counted using countBarcodedReads()
and then saved to a colData column named using the IDs. The
.bam file to use is specified by its filepath. Here we
specify the probe that were interested in is the “gRNA”
probe, which points the function to the probe ID stored in the
grnaProbe slot of the TapestriExperiment
object. Specifying “barcode” similarly points to the probe ID
barcodeProbe slot.
To account for PCR errors, the function by default allows for 2 base
mismatches. This can be altered to the user’s preference. Indels can be
allowed as well using with.indels = TRUE but is set to
FALSE by default.
grnaProbe(tapexp)
#> [1] "CO610_AMP350"
barcodeProbe(tapexp)
#> [1] "CO610_AMP351"
tapexp <- countBarcodedReads(tapexp, bam.file = "./tapexp.bam", probe = "grna", barcode.lookup = grna.lookup,
max.mismatch = 2, with.indels = F)The counts for the two gRNA sequences in each cell are now associated with the corresponding cell barcode.
colData(tapexp)
#> DataFrame with 1825 rows and 5 columns
#> cell.barcode total.reads cluster gRNA1 gRNA2
#> <character> <numeric> <factor> <numeric> <numeric>
#> AACAACCTACAATGTGCT-1 AACAACCTACAATGTGCT-1 27595 RPE1 18 0
#> AACAACTGGCGACCATCA-1 AACAACTGGCGACCATCA-1 33325 HCEC 0 66
#> AACAATGCAAGGATGCGT-1 AACAATGCAAGGATGCGT-1 36978 HCEC 0 0
#> AACAATGCATCGACGTTG-1 AACAATGCATCGACGTTG-1 25756 HCEC 0 34
#> AACAATGCATGAGAATCC-1 AACAATGCATGAGAATCC-1 26980 HCEC 0 0
#> ... ... ... ... ... ...
#> TTGGACTTCTATCATGCT-1 TTGGACTTCTATCATGCT-1 37431 HCEC 0 0
#> TTGGAGAACACGCAAGAT-1 TTGGAGAACACGCAAGAT-1 17456 RPE1 2 0
#> TTGGTAACTACCACTAGG-1 TTGGTAACTACCACTAGG-1 69296 RPE1 150 0
#> TTGGTAACTCAATCTCCG-1 TTGGTAACTCAATCTCCG-1 17866 RPE1 5 0
#> TTGTCAACCAGACTTCGT-1 TTGTCAACCAGACTTCGT-1 31021 HPNE 0 0To quickly assign sample labels to the cells, you can use
callSampleLabels() to assign a label based on the gRNA
counts. This creates a colData entry specified by
output.feature and labels the cell with whichever of the
input.features has the highest number of counts. If there
are no counts for any input feature, the return value is
NA. See the documentation for ways to customize the
parameters of this process.
tapexp <- callSampleLables(tapexp, input.features = c("gRNA1", "gRNA2"), output.feature = "sample.id")
colData(tapexp)
#> DataFrame with 1825 rows and 6 columns
#> cell.barcode total.reads cluster gRNA1 gRNA2 sample.id
#> <character> <numeric> <factor> <numeric> <numeric> <factor>
#> AACAACCTACAATGTGCT-1 AACAACCTACAATGTGCT-1 27595 RPE1 18 0 gRNA1
#> AACAACTGGCGACCATCA-1 AACAACTGGCGACCATCA-1 33325 HCEC 0 66 gRNA2
#> AACAATGCAAGGATGCGT-1 AACAATGCAAGGATGCGT-1 36978 HCEC 0 0 NA
#> AACAATGCATCGACGTTG-1 AACAATGCATCGACGTTG-1 25756 HCEC 0 34 gRNA2
#> AACAATGCATGAGAATCC-1 AACAATGCATGAGAATCC-1 26980 HCEC 0 0 NA
#> ... ... ... ... ... ... ...
#> TTGGACTTCTATCATGCT-1 TTGGACTTCTATCATGCT-1 37431 HCEC 0 0 NA
#> TTGGAGAACACGCAAGAT-1 TTGGAGAACACGCAAGAT-1 17456 RPE1 2 0 gRNA1
#> TTGGTAACTACCACTAGG-1 TTGGTAACTACCACTAGG-1 69296 RPE1 150 0 gRNA1
#> TTGGTAACTCAATCTCCG-1 TTGGTAACTCAATCTCCG-1 17866 RPE1 5 0 gRNA1
#> TTGTCAACCAGACTTCGT-1 TTGTCAACCAGACTTCGT-1 31021 HPNE 0 0 NAYou can visualize the results a few different ways, including as the annotations on a UMAP or heatmap. Here we show that each of the gRNAs were transduced in a specific cell line from the experiment.
reducedDimPlot(tapexp, dim.reduction = "umap", group.label = "sample.id")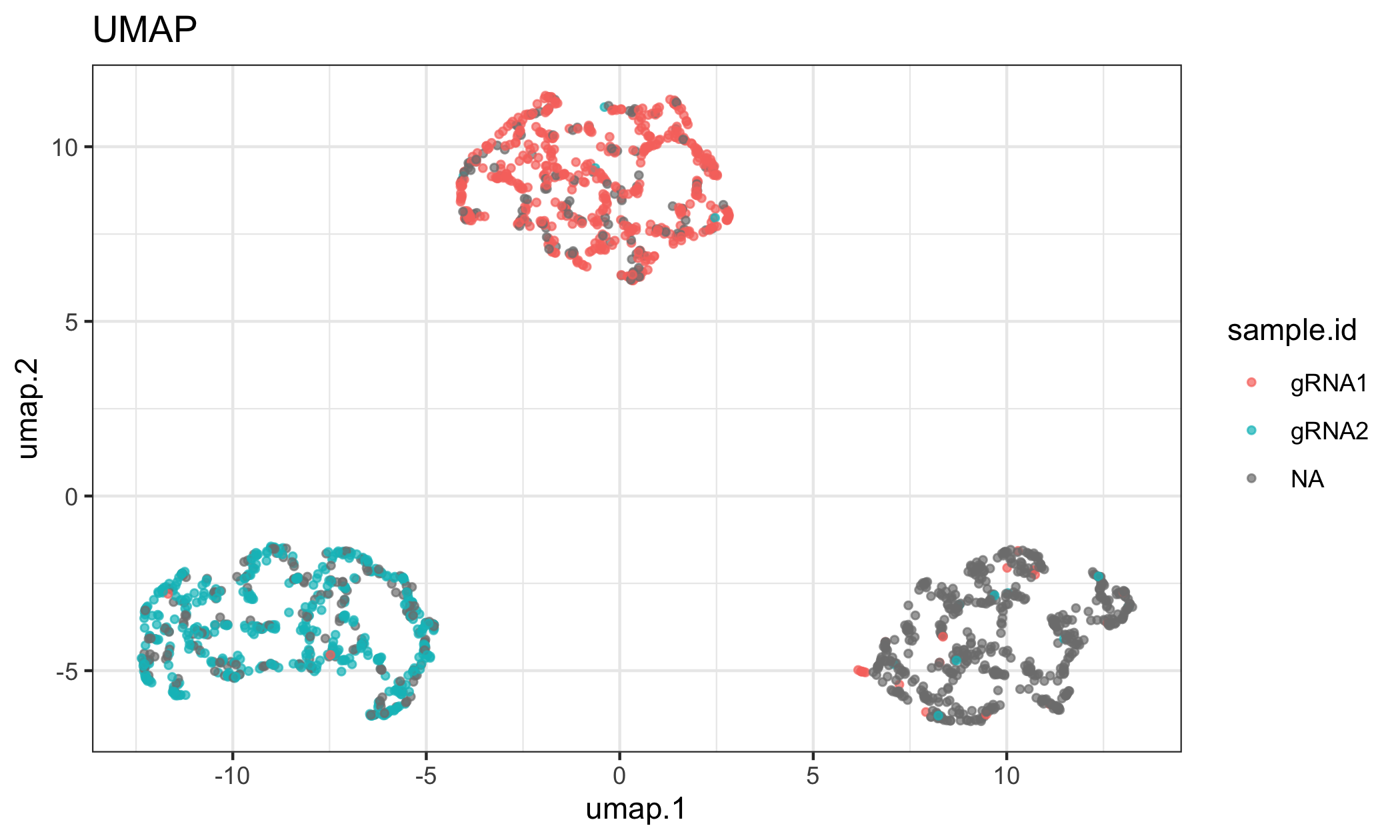
assayHeatmap(tapexp, alt.exp = "smoothedCopyNumberByChr", assay = "gmmCopyNumber",
split.row.by = "cluster", annotate.row.by = "sample.id", color.preset = "copy.number")
sessioninfo::session_info()
#> ─ Session info ───────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────
#> setting value
#> version R version 4.3.1 (2023-06-16)
#> os macOS Ventura 13.5
#> system x86_64, darwin20
#> ui RStudio
#> language (EN)
#> collate en_US.UTF-8
#> ctype en_US.UTF-8
#> tz America/New_York
#> date 2023-09-16
#> rstudio 2023.06.0+421 Mountain Hydrangea (desktop)
#> pandoc 3.1.1 @ /Applications/RStudio.app/Contents/Resources/app/quarto/bin/tools/ (via rmarkdown)
#>
#> ─ Packages ───────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────
#> ! package * version date (UTC) lib source
#> abind 1.4-5 2016-07-21 [1] CRAN (R 4.3.0)
#> askpass 1.2.0 2023-09-03 [1] CRAN (R 4.3.0)
#> Biobase * 2.60.0 2023-05-11 [1] Bioconductor
#> BiocGenerics * 0.46.0 2023-05-11 [1] Bioconductor
#> BiocParallel 1.34.2 2023-05-22 [1] Bioconductor
#> Biostrings 2.68.1 2023-05-16 [1] Bioconductor
#> bitops 1.0-7 2021-04-24 [1] CRAN (R 4.3.0)
#> brio 1.1.3 2021-11-30 [1] CRAN (R 4.3.0)
#> bslib 0.5.0 2023-06-09 [1] CRAN (R 4.3.0)
#> cachem 1.0.8 2023-05-01 [1] CRAN (R 4.3.0)
#> Cairo 1.6-0 2022-07-05 [1] CRAN (R 4.3.0)
#> callr 3.7.3 2022-11-02 [1] CRAN (R 4.3.0)
#> circlize 0.4.15 2022-05-10 [1] CRAN (R 4.3.0)
#> cli 3.6.1 2023-03-23 [1] CRAN (R 4.3.0)
#> clue 0.3-64 2023-01-31 [1] CRAN (R 4.3.0)
#> cluster 2.1.4 2022-08-22 [1] CRAN (R 4.3.1)
#> codetools 0.2-19 2023-02-01 [1] CRAN (R 4.3.1)
#> colorspace 2.1-0 2023-01-23 [1] CRAN (R 4.3.0)
#> commonmark 1.9.0 2023-03-17 [1] CRAN (R 4.3.0)
#> ComplexHeatmap 2.16.0 2023-05-11 [1] Bioconductor
#> cowplot 1.1.1 2020-12-30 [1] CRAN (R 4.3.0)
#> crayon 1.5.2 2022-09-29 [1] CRAN (R 4.3.0)
#> credentials 1.3.2 2021-11-29 [1] CRAN (R 4.3.0)
#> curl 5.0.2 2023-08-14 [1] CRAN (R 4.3.0)
#> dbscan 1.1-11 2022-10-27 [1] CRAN (R 4.3.0)
#> DelayedArray 0.26.7 2023-07-28 [1] Bioconductor
#> desc 1.4.2 2022-09-08 [1] CRAN (R 4.3.0)
#> devtools * 2.4.5 2022-10-11 [1] CRAN (R 4.3.0)
#> digest 0.6.33 2023-07-07 [1] CRAN (R 4.3.0)
#> distributional 0.3.2 2023-03-22 [1] CRAN (R 4.3.0)
#> doParallel 1.0.17 2022-02-07 [1] CRAN (R 4.3.0)
#> downlit 0.4.3 2023-06-29 [1] CRAN (R 4.3.0)
#> dplyr * 1.1.3 2023-09-03 [1] CRAN (R 4.3.0)
#> ellipsis 0.3.2 2021-04-29 [1] CRAN (R 4.3.0)
#> evaluate 0.21 2023-05-05 [1] CRAN (R 4.3.0)
#> fansi 1.0.4 2023-01-22 [1] CRAN (R 4.3.0)
#> farver 2.1.1 2022-07-06 [1] CRAN (R 4.3.0)
#> fastmap 1.1.1 2023-02-24 [1] CRAN (R 4.3.0)
#> fitdistrplus * 1.1-11 2023-04-25 [1] CRAN (R 4.3.0)
#> forcats * 1.0.0 2023-01-29 [1] CRAN (R 4.3.0)
#> foreach 1.5.2 2022-02-02 [1] CRAN (R 4.3.0)
#> formatR 1.14 2023-01-17 [1] CRAN (R 4.3.0)
#> fs 1.6.3 2023-07-20 [1] CRAN (R 4.3.0)
#> generics 0.1.3 2022-07-05 [1] CRAN (R 4.3.0)
#> GenomeInfoDb * 1.36.2 2023-08-25 [1] Bioconductor
#> GenomeInfoDbData 1.2.10 2023-07-09 [1] Bioconductor
#> GenomicRanges * 1.52.0 2023-05-11 [1] Bioconductor
#> gert 1.9.2 2022-12-05 [1] CRAN (R 4.3.0)
#> GetoptLong 1.0.5 2020-12-15 [1] CRAN (R 4.3.0)
#> ggdist * 3.3.0 2023-05-13 [1] CRAN (R 4.3.0)
#> ggplot2 * 3.4.3 2023-08-14 [1] CRAN (R 4.3.0)
#> ggrepel 0.9.3 2023-02-03 [1] CRAN (R 4.3.0)
#> ggridges 0.5.4 2022-09-26 [1] CRAN (R 4.3.0)
#> gh 1.4.0 2023-02-22 [1] CRAN (R 4.3.0)
#> gitcreds 0.1.2 2022-09-08 [1] CRAN (R 4.3.0)
#> GlobalOptions 0.1.2 2020-06-10 [1] CRAN (R 4.3.0)
#> glue 1.6.2 2022-02-24 [1] CRAN (R 4.3.0)
#> gtable 0.3.4 2023-08-21 [1] CRAN (R 4.3.0)
#> gtools 3.9.4 2022-11-27 [1] CRAN (R 4.3.0)
#> here 1.0.1 2020-12-13 [1] CRAN (R 4.3.0)
#> highr 0.10 2022-12-22 [1] CRAN (R 4.3.0)
#> htmltools 0.5.5 2023-03-23 [1] CRAN (R 4.3.0)
#> htmlwidgets 1.6.2 2023-03-17 [1] CRAN (R 4.3.0)
#> httpuv 1.6.11 2023-05-11 [1] CRAN (R 4.3.0)
#> httr2 0.2.3 2023-05-08 [1] CRAN (R 4.3.0)
#> IRanges * 2.34.1 2023-06-22 [1] Bioconductor
#> iterators 1.0.14 2022-02-05 [1] CRAN (R 4.3.0)
#> janitor 2.2.0 2023-02-02 [1] CRAN (R 4.3.0)
#> jquerylib 0.1.4 2021-04-26 [1] CRAN (R 4.3.0)
#> jsonlite 1.8.7 2023-06-29 [1] CRAN (R 4.3.0)
#> VP karyotapR * 1.0.1.9000 2023-09-07 [?] CRAN (R 4.3.1) (on disk 1.0.1)
#> knitr 1.43 2023-05-25 [1] CRAN (R 4.3.0)
#> labeling 0.4.3 2023-08-29 [1] CRAN (R 4.3.0)
#> later 1.3.1 2023-05-02 [1] CRAN (R 4.3.0)
#> lattice 0.21-8 2023-04-05 [1] CRAN (R 4.3.1)
#> lifecycle 1.0.3 2022-10-07 [1] CRAN (R 4.3.0)
#> lubridate 1.9.2 2023-02-10 [1] CRAN (R 4.3.0)
#> magick 2.7.4 2023-03-09 [1] CRAN (R 4.3.0)
#> magrittr 2.0.3 2022-03-30 [1] CRAN (R 4.3.0)
#> MASS * 7.3-60 2023-05-04 [1] CRAN (R 4.3.1)
#> Matrix 1.6-0 2023-07-08 [1] CRAN (R 4.3.0)
#> MatrixGenerics * 1.12.3 2023-07-31 [1] Bioconductor
#> matrixStats * 1.0.0 2023-06-02 [1] CRAN (R 4.3.0)
#> memoise 2.0.1 2021-11-26 [1] CRAN (R 4.3.0)
#> mime 0.12 2021-09-28 [1] CRAN (R 4.3.0)
#> miniUI 0.1.1.1 2018-05-18 [1] CRAN (R 4.3.0)
#> munsell 0.5.0 2018-06-12 [1] CRAN (R 4.3.0)
#> openssl 2.1.0 2023-07-15 [1] CRAN (R 4.3.0)
#> pillar 1.9.0 2023-03-22 [1] CRAN (R 4.3.0)
#> pkgbuild 1.4.2 2023-06-26 [1] CRAN (R 4.3.0)
#> pkgconfig 2.0.3 2019-09-22 [1] CRAN (R 4.3.0)
#> pkgdown 2.0.7 2022-12-14 [1] CRAN (R 4.3.0)
#> pkgload 1.3.2.1 2023-07-08 [1] CRAN (R 4.3.0)
#> png 0.1-8 2022-11-29 [1] CRAN (R 4.3.0)
#> prettyunits 1.1.1 2020-01-24 [1] CRAN (R 4.3.0)
#> processx 3.8.2 2023-06-30 [1] CRAN (R 4.3.0)
#> profvis 0.3.8 2023-05-02 [1] CRAN (R 4.3.0)
#> promises 1.2.0.1 2021-02-11 [1] CRAN (R 4.3.0)
#> ps 1.7.5 2023-04-18 [1] CRAN (R 4.3.0)
#> purrr * 1.0.2 2023-08-10 [1] CRAN (R 4.3.0)
#> R6 2.5.1 2021-08-19 [1] CRAN (R 4.3.0)
#> rappdirs 0.3.3 2021-01-31 [1] CRAN (R 4.3.0)
#> rcmdcheck 1.4.0 2021-09-27 [1] CRAN (R 4.3.0)
#> RColorBrewer 1.1-3 2022-04-03 [1] CRAN (R 4.3.0)
#> Rcpp 1.0.11 2023-07-06 [1] CRAN (R 4.3.0)
#> RCurl 1.98-1.12 2023-03-27 [1] CRAN (R 4.3.0)
#> remotes 2.4.2.1 2023-07-18 [1] CRAN (R 4.3.0)
#> reticulate 1.31 2023-08-10 [1] CRAN (R 4.3.0)
#> rhdf5 2.44.0 2023-05-11 [1] Bioconductor
#> rhdf5filters 1.12.1 2023-05-11 [1] Bioconductor
#> Rhdf5lib 1.22.0 2023-05-11 [1] Bioconductor
#> rjson 0.2.21 2022-01-09 [1] CRAN (R 4.3.0)
#> rlang 1.1.1 2023-04-28 [1] CRAN (R 4.3.0)
#> rmarkdown 2.23 2023-07-01 [1] CRAN (R 4.3.0)
#> roxygen2 7.2.3 2022-12-08 [1] CRAN (R 4.3.0)
#> rprojroot 2.0.3 2022-04-02 [1] CRAN (R 4.3.0)
#> Rsamtools 2.16.0 2023-05-11 [1] Bioconductor
#> RSpectra 0.16-1 2022-04-24 [1] CRAN (R 4.3.0)
#> rstudioapi 0.15.0 2023-07-07 [1] CRAN (R 4.3.0)
#> S4Arrays 1.0.6 2023-08-30 [1] Bioconductor
#> S4Vectors * 0.38.1 2023-05-11 [1] Bioconductor
#> sass 0.4.7 2023-07-15 [1] CRAN (R 4.3.0)
#> scales 1.2.1 2022-08-20 [1] CRAN (R 4.3.0)
#> sessioninfo 1.2.2 2021-12-06 [1] CRAN (R 4.3.0)
#> shape 1.4.6 2021-05-19 [1] CRAN (R 4.3.0)
#> shiny 1.7.4.1 2023-07-06 [1] CRAN (R 4.3.0)
#> SingleCellExperiment * 1.22.0 2023-05-11 [1] Bioconductor
#> snakecase 0.11.0 2019-05-25 [1] CRAN (R 4.3.0)
#> stringdist 0.9.10 2022-11-07 [1] CRAN (R 4.3.0)
#> stringi 1.7.12 2023-01-11 [1] CRAN (R 4.3.0)
#> stringr 1.5.0 2022-12-02 [1] CRAN (R 4.3.0)
#> SummarizedExperiment * 1.30.2 2023-06-06 [1] Bioconductor
#> survival * 3.5-5 2023-03-12 [1] CRAN (R 4.3.1)
#> sys 3.4.2 2023-05-23 [1] CRAN (R 4.3.0)
#> testthat 3.1.10 2023-07-06 [1] CRAN (R 4.3.0)
#> tibble 3.2.1 2023-03-20 [1] CRAN (R 4.3.0)
#> tidyr 1.3.0 2023-01-24 [1] CRAN (R 4.3.0)
#> tidyselect 1.2.0 2022-10-10 [1] CRAN (R 4.3.0)
#> timechange 0.2.0 2023-01-11 [1] CRAN (R 4.3.0)
#> umap 0.2.10.0 2023-02-01 [1] CRAN (R 4.3.0)
#> urlchecker 1.0.1 2021-11-30 [1] CRAN (R 4.3.0)
#> usethis * 2.2.2 2023-07-06 [1] CRAN (R 4.3.0)
#> utf8 1.2.3 2023-01-31 [1] CRAN (R 4.3.0)
#> vctrs 0.6.3 2023-06-14 [1] CRAN (R 4.3.0)
#> viridisLite 0.4.2 2023-05-02 [1] CRAN (R 4.3.0)
#> whisker 0.4.1 2022-12-05 [1] CRAN (R 4.3.0)
#> withr 2.5.0 2022-03-03 [1] CRAN (R 4.3.0)
#> xfun 0.39 2023-04-20 [1] CRAN (R 4.3.0)
#> xml2 1.3.5 2023-07-06 [1] CRAN (R 4.3.0)
#> xopen 1.0.0 2018-09-17 [1] CRAN (R 4.3.0)
#> xtable 1.8-4 2019-04-21 [1] CRAN (R 4.3.0)
#> XVector 0.40.0 2023-05-11 [1] Bioconductor
#> yaml 2.3.7 2023-01-23 [1] CRAN (R 4.3.0)
#> zlibbioc 1.46.0 2023-05-11 [1] Bioconductor
#>
#> [1] /Library/Frameworks/R.framework/Versions/4.3-x86_64/Resources/library
#>
#> V ── Loaded and on-disk version mismatch.
#> P ── Loaded and on-disk path mismatch.
#>
#> ──────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────